Research rDNAome - GANLEY Laboratory
Research
Our lab is primarily interested in two kinds of rDNA: ribosomal DNA and random DNA. The backdrop to these is that both offer opportunities for better understanding the evolution of eukaryote genomes, albeit in very different ways. We use a variety of model systems to investigate these two kinds of rDNA, but principally these are the brewing yeast Saccharomyces cerevisiae and human cell lines. We investigate these rDNAs using both molecular biology and computational approaches. We also maintain interests in fungal genome evolution, polyploidy, human genetics, and mushrooms.
Introduction
Current research projects
- Evolutionary dynamics of ribosomal RNA gene repeats
- Function of the TAR1 gene
- Random DNA and the junk vs dark DNA debate
Organisation and structure of the ribosomal RNA gene repeats (rDNA)
The rDNA is present as head-to-tail tandem repeats in most eukaryotes (Figure 1), and this organization appears to have been present in the last common ancestor of eukaryotes. A single rDNA repeat unit can range in size from ~8-45kb, depending on the organism. It encodes the major species of ribosomal RNA that make up the ribosome and the rDNA repeats are found within the nucleolus, the site of primary ribosome biogenesis. rDNA repeat arrays may be present at one-or-more chromosomal locations. The rDNA is an essential and ancient component of genomes.
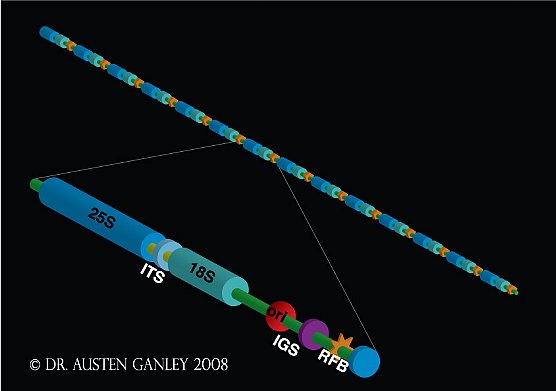
Figure 1: Organisation of the ribosomal DNA repeats in eukaryotes.
A single repeat unit consists of the 18S, 5.8S and 28S ribosomal RNA genes separated by internal transcribed spacers (ITS) 1 and 2. Between adjacent coding regions is a large spacer region, the intergenic spacer (IGS). In some species, the IGS harbours another ribosomal RNA gene, the 5S rRNA. Although detailed IGS characterization has been performed for few organisms, an origin of replication (ori) and replication fork barrier site (RFB) have been found in the IGS of organisms that have been examined.
The repetitive nature of the rDNA makes it prone to unequal recombination between repeats, resulting in copy number variation, and there is an extensive regulation system in place to control rDNA recombination. Therefore, while copy number in a species is held roughly constant, the precise copy number varies between individuals due to recombination-based copy number fluctuation. This recombination has a powerful influence on the evolution of the rDNA repeats. It results in the rDNA evolving via the evolutionary pattern of concerted evolution that is characterized by a high level of sequence identity between the repeats within a genome. In the yeast, Saccharomyces cerevisiae, we have shown that non-coding transcription from a bidirectional promoter in the rDNA, called E-pro, is a key regulator of rDNA recombination, with the level of this noncoding transcription being modulated by the silencing gene, SIR2. This recombination is also dependent on the fork blocking protein, Fob1p, which binds to the replication fork barrier (RFB) site in the rDNA. Binding of Fob1p to the RFB blocks the replication fork in one direction, and induces double strand break formation that is repaired by homologous recombination with another repeat. This recombination appears to depend on replication, and each rDNA unit contains a replication origin. Therefore replication initiates within the rDNA, although origins only fire in a subset of rDNA repeat units in each cell cycle.
The rDNA is also an extremely interesting epigenetic system, as although there are a large number of rDNA copies with very similar DNA sequences, at least in most conditions only a subset (half or less) are actually being transcribed. For those repeat copies that are transcribed (by RNA polymerase I), the transcription level is the highest of any gene in the genome, and this high level of transcription influences the behaviour of the rDNA. However, the remaining repeats are completely transcriptionally silent. We have shown that rDNA units within a genome are all essentially identical at the DNA sequence level, therefore this mosaic pattern of active/inactive units must be determined epigenetically. There is great interest in understanding how this occurs, as rRNA transcription level correlates with growth rate, and many cancer cells increase the number of active rDNA units they have.
Evolutionary dynamics of the ribosomal RNA gene repeats
The rDNA repeats are a fascinating and unique evolutionary system. The ongoing recombination-based duplication and deletion of repeat copies drives concerted evolution, yet we only understand how this work in a general sense. Therefore, we are interested in better describing the variation between rDNA repeats, which is a consequence of these copy number dynamics and mutation. However, while evolution is occurring between repeats in arrays, it is also occurring at the level of the organism, exemplified by the rDNA containing both highly conserved regions (the rRNA genes) and extremely variable regions. The repeat dynamics occurring within the rDNA are therefore partly a form of multilevel selection conflict between the interests of the repeat (repeats that can replicate themselves irrespective of their ability to encode rRNA are favoured at one level) and the interests of the host (which requires repeats that encode rRNA). We want to better understand the nature of this conflict and how it is resolved. Adding to this is yet another level of multi-level selection dynamics – small tandem repeats within single rDNA units that undergo their own copy number dynamics. We are also interested in elucidating the molecular mechanisms that underpin the copy number dynamics of the rDNA repeats and repeats contained within them, as well as the regulation of copy number change.
Function of the TAR1 gene
Hemiascomycete yeasts curiously encode a gene, known as TAR1, that is located antisense to the large ribosomal RNA subunit gene in the rDNA. This gene is therefore multi-copy, yet is expressed at very low levels. It has evolved de novo, from DNA sequence that was previously only used to encode rRNA on the opposite strand. The protein it encodes, Tar1p, is localized to mitochondria, but the role of Tar1p remains unclear. We have speculated on what the function of this unusual gene might be and how it ties into the multicopy nature of the rDNA, and we are interested in experimentally testing these ideas.
Random DNA and the junk vs dark DNA debate
 Only a tiny proportion of large eukaryote genomes such as the human genome consists of genes. The nature of much of the remaining parts of the genome has been debated for decades, with the debate typically occurring between two camps. One posits that the majority of the genomes consists of uncharacterized but functional ‘dark’ DNA. The other posits that the majority of these genomes consist of unused and unnecessary ‘junk’ DNA. A key observation that has been used in this debate is that most of the DNA contained within these genomes exhibits biochemical activities, such as transcription. Are these ‘pervasive genomic activities’ traces of underlying functional activities, which must therefore be widespread in genomes? Or are they simply a consequence of background noise? Distinguishing between these alternatives is a key step in resolving the dark vs junk DNA debate, and a way to do this was first proposed by Sean Eddy in 2013: determine whether completely random DNA that is inserted into genomes shows pervasive genomic activities or not.
Only a tiny proportion of large eukaryote genomes such as the human genome consists of genes. The nature of much of the remaining parts of the genome has been debated for decades, with the debate typically occurring between two camps. One posits that the majority of the genomes consists of uncharacterized but functional ‘dark’ DNA. The other posits that the majority of these genomes consist of unused and unnecessary ‘junk’ DNA. A key observation that has been used in this debate is that most of the DNA contained within these genomes exhibits biochemical activities, such as transcription. Are these ‘pervasive genomic activities’ traces of underlying functional activities, which must therefore be widespread in genomes? Or are they simply a consequence of background noise? Distinguishing between these alternatives is a key step in resolving the dark vs junk DNA debate, and a way to do this was first proposed by Sean Eddy in 2013: determine whether completely random DNA that is inserted into genomes shows pervasive genomic activities or not.
We have taken up Eddy’s challenge, and to do so we have pioneered an enzymatic method for generating large pieces of random DNA. We are stitching these random DNA fragments into larger, chromosomal-sized pieces of DNA for insertion into the genomes of organisms. Once this is done, we will measure the genomic activities of these random DNA chromosomes. If the random chromosomes exhibit similar genomic activities to the noncoding parts of genomes, this would suggest that background noise explains much of the pervasive genomic activities, thus these activities provide no particular evidence for function. Conversely, if genomic activities are suppressed in these random chromosomes, this would suggest that pervasive genomic activities in the noncoding regions of genomes are there as a result of selection.